
There are many tools for generating genome assemblies. Each tool has different properties and performs differently on different datasets. Therefore, one important task in genetics is comparing genome assemblies in order to know what software performs better in every circumstance.
For assembling genomes from samples designed to capture a specific genome (cultivating the organism, taking a tissue sample directly from the organism, or capturing specific sequences by library design), the comparison of results tends to be an easy concept. In case you have a reference genome you just map your assembly to the reference and you get a quite clear idea of how has the assembly performed. This is a technique not free of errors and inconsistencies but in general works relatively well. A tool frequently used for this task is QUAST.
In case you don’t have a reference sequence, or also in case you have it but you want to get another measure of completeness, you can try to identify genes that are usually present in organisms of the family of the organism that you are sequencing. A tool that takes a measure of completeness this way is BUSCO.
But metagenomes and transcriptomes belong to a different case. Now, you don’t know exactly what you are sequencing. In the case of transcriptomes, you are looking for thousands of different mRNA sequences, and despite sometimes you have references, these references are a work in progress. Also, when you assemble a transcriptome you are quite often looking for a mutation, for example in cancer transcriptome samples. These mutations often are chimeras from two genes, or from a gene and a transposable element. Therefore, you can’t just rely on references. In the cast of metagenomes, you don’t even know what you are looking for. There are potentially hundreds of microorganisms in the sample and your goal is to find the largest genome sequence from all of them. Often, these are even new species that have never been sequenced. So, you can’t either rely on references in this case.
For cases in which you can’t rely on references a common method for evaluating the performance of the assembly consists on mapping the reads to every assembly. The assembly to which more percentage of reads map is therefore understood to be the one that did a better job finding a sequence for each read. But that is not necessarily true or accurate. One assembly could have thousands of short contigs to which a large percentage of reads map but still be useless for biological studies.
This week I have been working on a different approach. Instead of mapping reads to each assembly, just map contigs from one assembly to the other one. As long as I know that is not much in use (if anyone does indeed use it) as a method for comparing assemblies, and yet I find it to be quite useful.
Here for example there is a mapping of some contigs in one transcriptomic assembly to a contig in another assembly.
k141_81226 6238 62 762 - 42703__length_700 700 0 700 693 700 32 NM:i:7 ms:i:1358 AS:i:1358 nn:i:0 tp:A:P cm:i:115 s1:i:631 s2:i:621 de:f:0.0100 rl:i:26 cg:Z:700M k141_81226 6238 111 762 + 4805__length_651 651 0 651 649 651 0 NM:i:2 ms:i:1290 AS:i:1290 nn:i:0 tp:A:S cm:i:114 s1:i:621 de:f:0.0031 rl:i:26 cg:Z:651M k141_81226 6238 2951 3396 + 6117__length_445 445 0 445 445 445 60 NM:i:0 ms:i:890 AS:i:890 nn:i:0 tp:A:P cm:i:78 s1:i:436 s2:i:0 de:f:0 rl:i:26 cg:Z:445M k141_81226 6238 4936 5334 - 32681__length_398 398 0 398 398 398 60 NM:i:0 ms:i:796 AS:i:796 nn:i:0 tp:A:P cm:i:68 s1:i:383 s2:i:0 de:f:0 rl:i:26 cg:Z:398M k141_81226 6238 2660 2985 - 56442__length_325 325 0 325 324 325 60 NM:i:1 ms:i:644 AS:i:644 nn:i:0 tp:A:P cm:i:54 s1:i:297 s2:i:0 de:f:0.0031 rl:i:26 cg:Z:325M k141_81226 6238 1891 2202 + 55945__length_312 312 0 312 311 312 35 NM:i:1 ms:i:616 AS:i:616 nn:i:0 tp:A:P cm:i:55 s1:i:284 s2:i:274 de:f:0.0032 rl:i:26 cg:Z:299M1D12M k141_81226 6238 1898 2202 - 36701__length_308 308 0 308 303 308 0 NM:i:5 ms:i:586 AS:i:586 nn:i:0 tp:A:S cm:i:53 s1:i:274 de:f:0.0098 rl:i:26 cg:Z:11M3D2M1D291M k141_81226 6238 5369 5556 + 23817__length_187 187 0 187 187 187 60 NM:i:0 ms:i:374 AS:i:374 nn:i:0 tp:A:P cm:i:27 s1:i:179 s2:i:0 de:f:0 rl:i:26 cg:Z:187M k141_81226 6238 5562 5737 - 10842__length_175 175 0 175 175 175 60 NM:i:0 ms:i:350 AS:i:350 nn:i:0 tp:A:P cm:i:32 s1:i:167 s2:i:0 de:f:0 rl:i:26 cg:Z:175M k141_81226 6238 5790 5940 - 37907__length_150 150 0 150 150 150 60 NM:i:0 ms:i:300 AS:i:300 nn:i:0 tp:A:P cm:i:19 s1:i:136 s2:i:0 de:f:0 rl:i:26 cg:Z:150M k141_81226 6238 2368 2518 - 2143__length_150 150 0 150 149 150 60 NM:i:1 ms:i:294 AS:i:294 nn:i:0 tp:A:P cm:i:23 s1:i:144 s2:i:0 de:f:0.0067 rl:i:26 cg:Z:150M
Find the PAF format description if you want to understand this table. But it basically says which contig matches whitch other contig and at which positions.
We can see how one large contig (6238 bps) maps to a few shorter contigs in the other assembly (none larger than 1000bps). We can understand that in this case the first assembly most likely did a better job and the second one just failed to assemble the sequence. But it could indeed be exactly the opposite. It could be that the first assembly got a misassembly merging a few mRNAs.
There are a few ways to check which case is the right one. You can, for example, use BLAST to find matches in an mRNA database. In that case you will find that k141_81226 matches "Homo sapiens SIN3 transcription regulator family member B (SIN3B), transcript variant 3, mRNA" at 66% identity, while the other contigs match in most cases to the same sequence but in smaller fraction. That seems already quite indicative of what has went on with these assemblies. It seems highly likely that the smaller contigs are just incomplete contigs and do not represent real mRNA sequences present in the sample, but we also find that the large sequence could in addition be a misassembly.
Even if you don’t have references or you find it unpractical to make a BLAST search for every contig, there is another way to get clues on what has gone on. You can make a multiple alignment of all the contigs. Then you can visualize the result and deduce what is a misassembly and what is just legit larger contigs.
This is what you get when the reads are incomplete sequences.
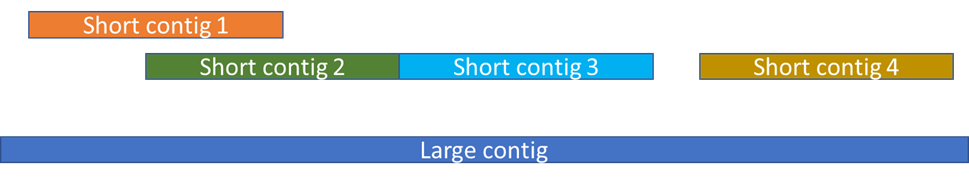
This is what you typically get when the large read is a misassembly.

And this is what you get when both assemblies are complementary and capture different regions of the same sequence.

The last case is, indeed, under my experience, the most frequent, even if one assembly gets larger and more numerous contigs. Therefore you usually can complement your assembly even with an assembly of lower quality.
I guess I should write a short paper with more details on this method and even develop a more robust tool, but I have no time and no resources to start side projects. Which in my opinion is a shame for the actual organization of science. Funds should go to where there is a demonstrated new idea with potential and not to where there is reputation and influences. One of the many things that need to be changed in science and innovation organization. Of course, getting private capital for such a project isn't either ever going to happen. Things are really so bad. No wonder why the promised future at last was not flying cars but... impotence to fight a pandemic.