We will show here a case of use with real data.
You can download the data from here and test yourself how s-aligner works. The data corresponds to the run SRR12956185. This corresponds to an Unbiased Deep Sequencing with Nextera XT on Illumina MiSeq for an African-lineage Zika virus.
 Download data
Download data
:~$ ./index.sh raw-zika-sample1 PATH_TO_UNCOMPRESSED_ZIP/sra_data.fasta
:~$ ./assemble.sh raw-zika-sample1 500 > results/raw-zika-sample1-500.fa
You will get an output in you terminal like this:
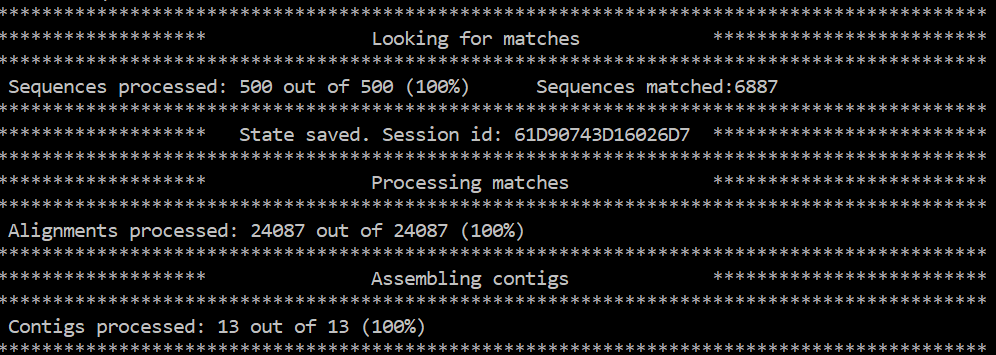
You can do that in many ways. For example you could use the Quast tool and the reference genome. A simpler way and often more informative is just making an online Blast search in the NCBI portal and see if the sequence matches a zika virus.
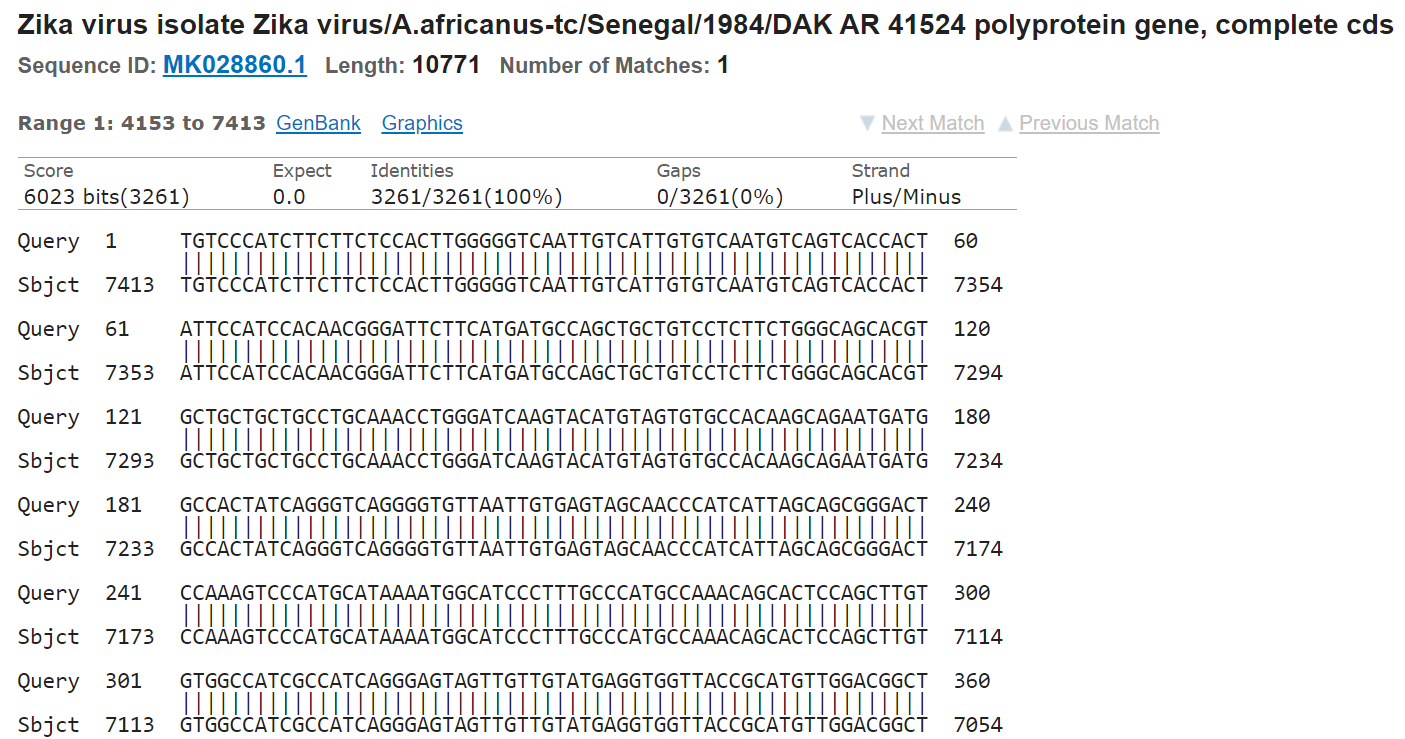
In this case, we see how the contig we introduced (3261 bp long) is a perfect match for the sequence MK028860.1, corresponding to a Zika virus assembly. So, we are in the good direction.
:~$ ./assemble.sh raw-zika-sample1 2500 -sid THE_SESSION_ID_IN_PREVIOUS_STEP > results/raw-zika-sample1-2500.fa
Important: Note that every time you run s-aligner with the same data you will likely get different results. In some cases even very different results. That's due to the randomness introduced by the order variation caused by multithreading.
Your quast result, in any case should look very similar to that:
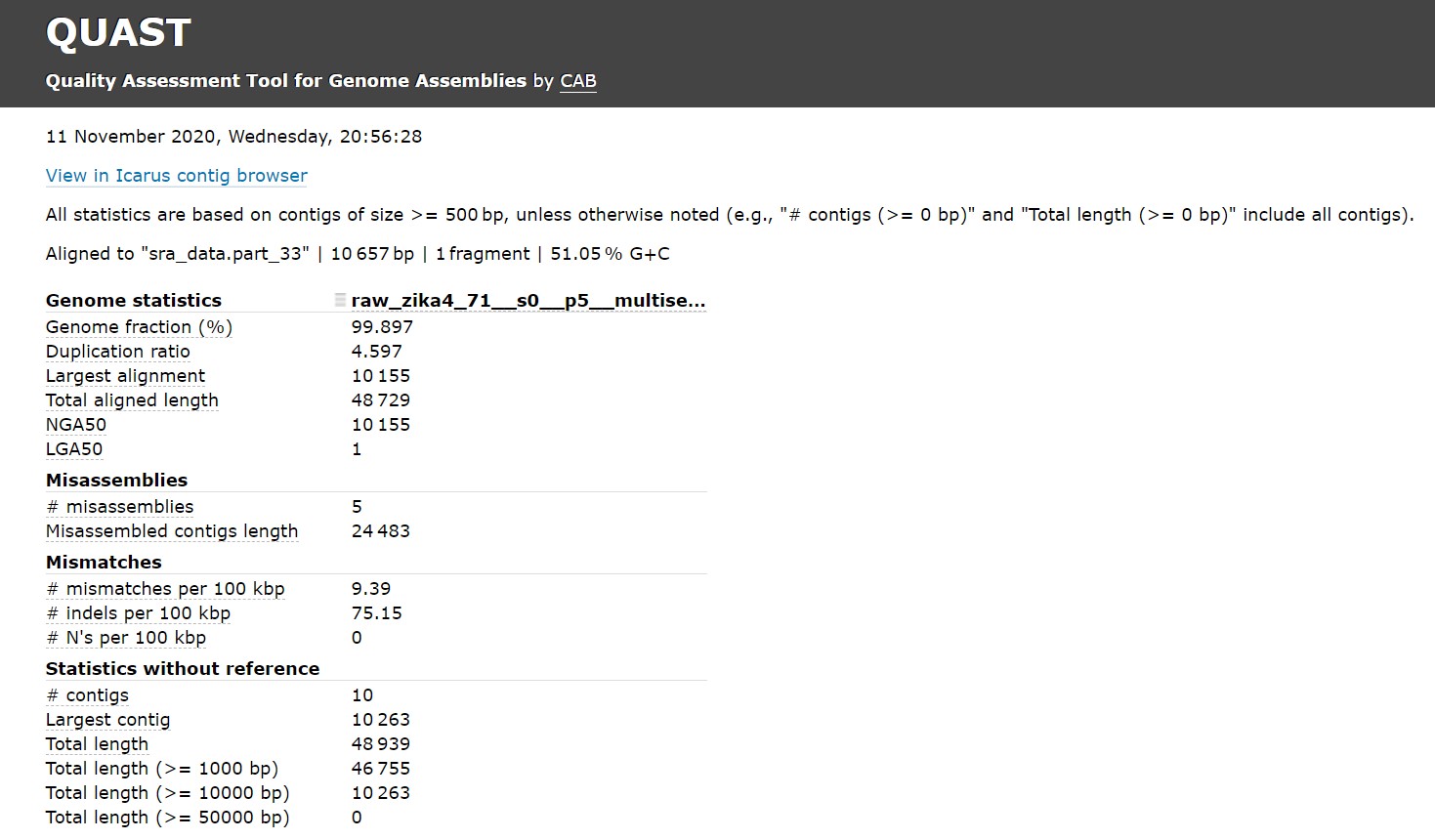
You can compare these results with results from other software tools. You can use the compressed fastq file in the zip that you downloaded as input for the other tools.
| LGA50 | NGA50 | Genome fraction (%) | Max contig length | Largest alignment | Misassembled length | |
|---|---|---|---|---|---|---|
| Velvet | it doesn't contain contigs >= 500 bp | |||||
| SPAdes | - | - | 0,72 | 880 | 77 | 0 |
| rnaSPAdes | 3 | 1.642 | 98,04 | 4.070 | 2.848 | 33.248 |
| s-aligner | 1 | 10.155 | 99,90 | 10.263 | 10.155 | 24.483 |