
Today marks a small milestone in the development of this project. A technical description of s-aligner containing a further analysis of its capabilities is finally available to the public. I hope it can be peer-reviewed in the next weeks or months. It is already being scrutinized by the community. I invite you to read it and participate in the discussion.
"s-aligner: a greedy algorithm for non-greedy denovo genome assembly"


If you don’t have much time, here you have also a summary of the main ideas and results.
S-aligner is a simple idea. No big breakthrough discovery has been required to develop it. Just high skills developing software and selecting/discarding technologies and ideas.
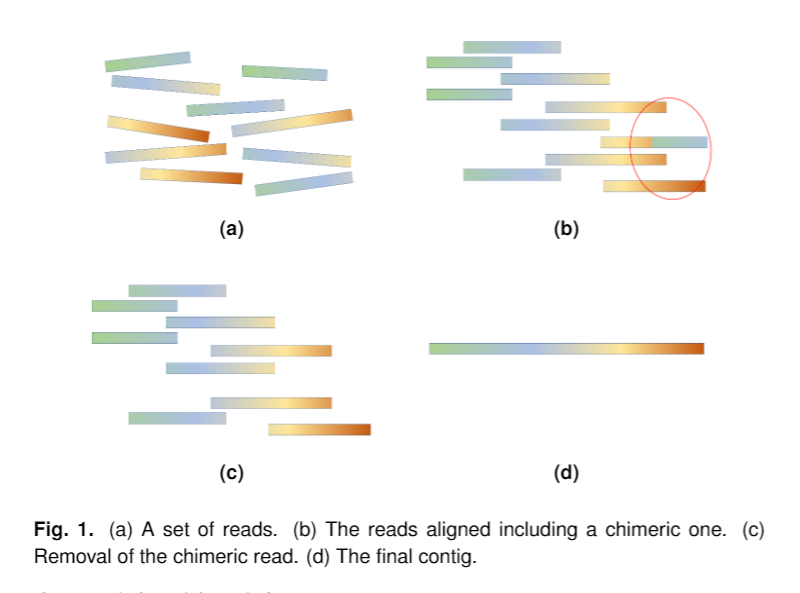
Some characteristics of s-aligner are:
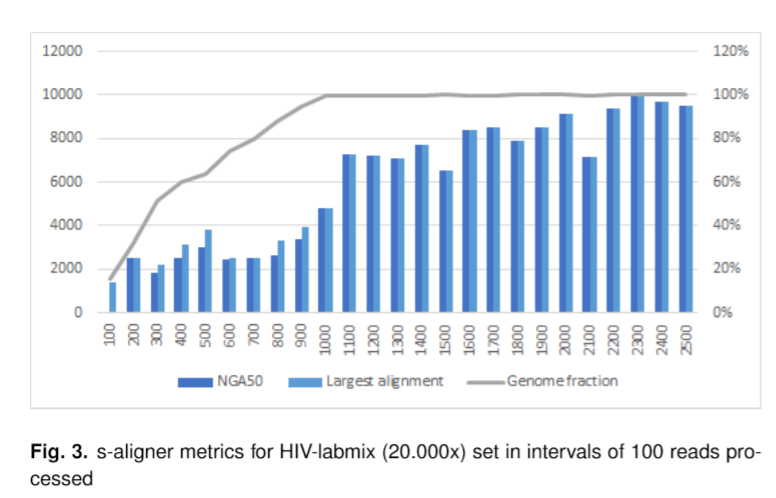
And it outperforms every software it was tested against for viral de novo genome assembly.
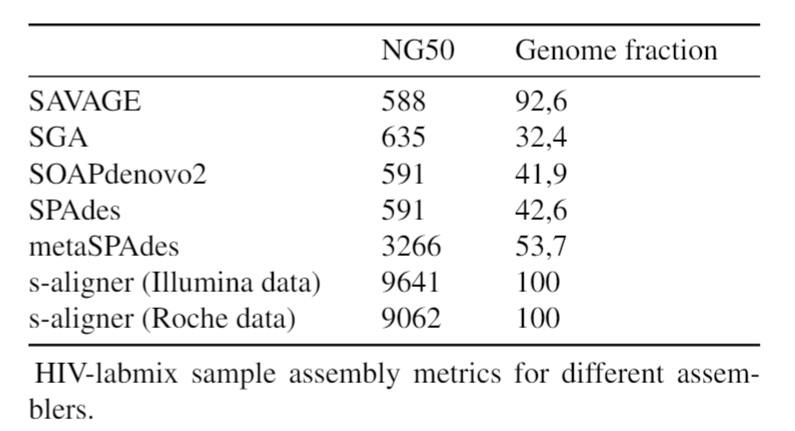
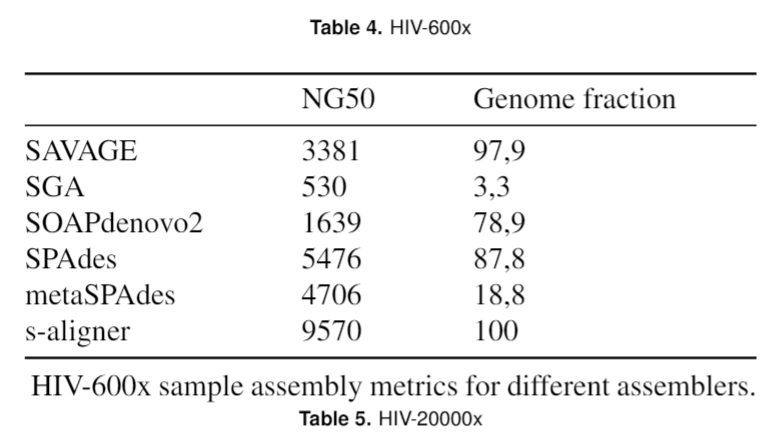
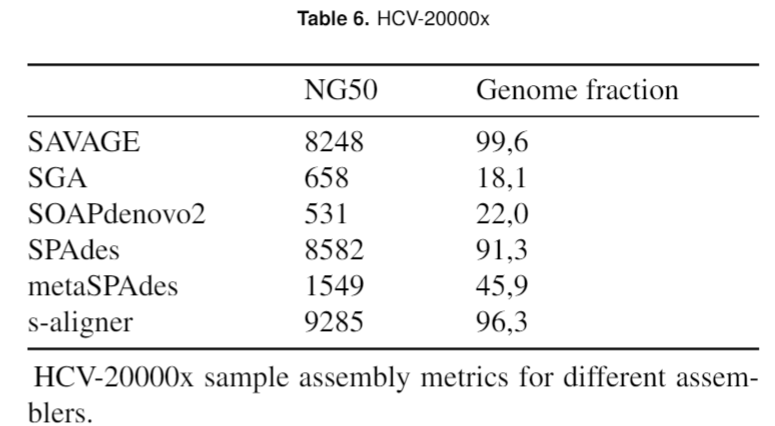
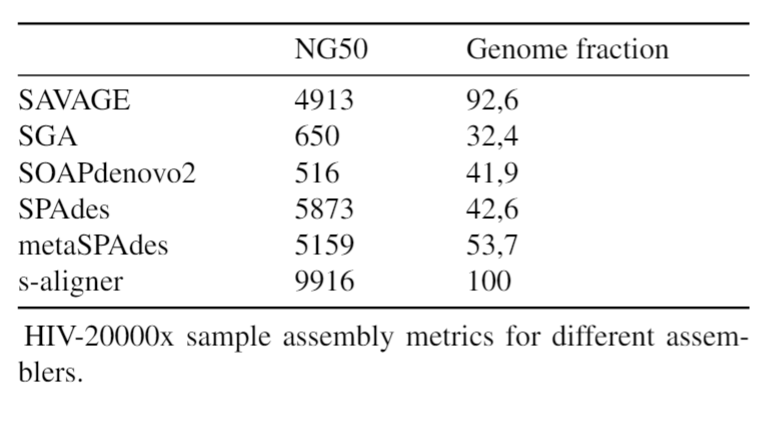
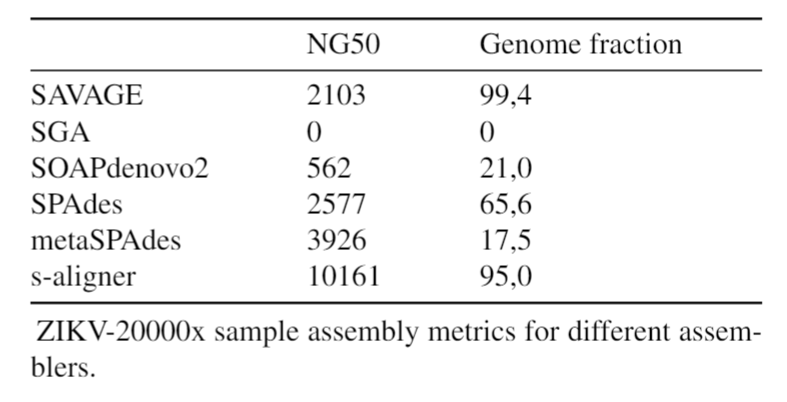
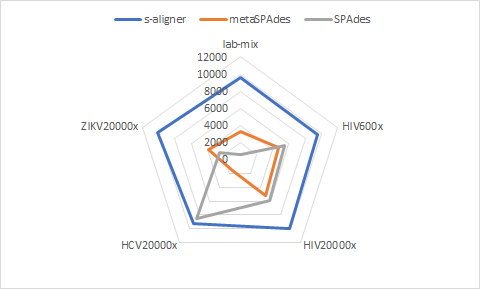
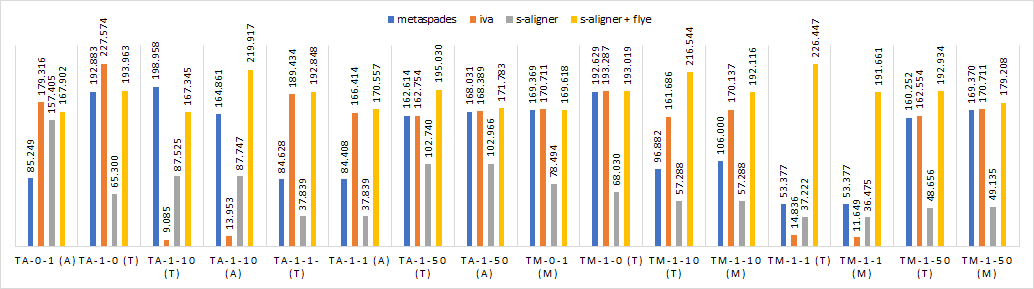
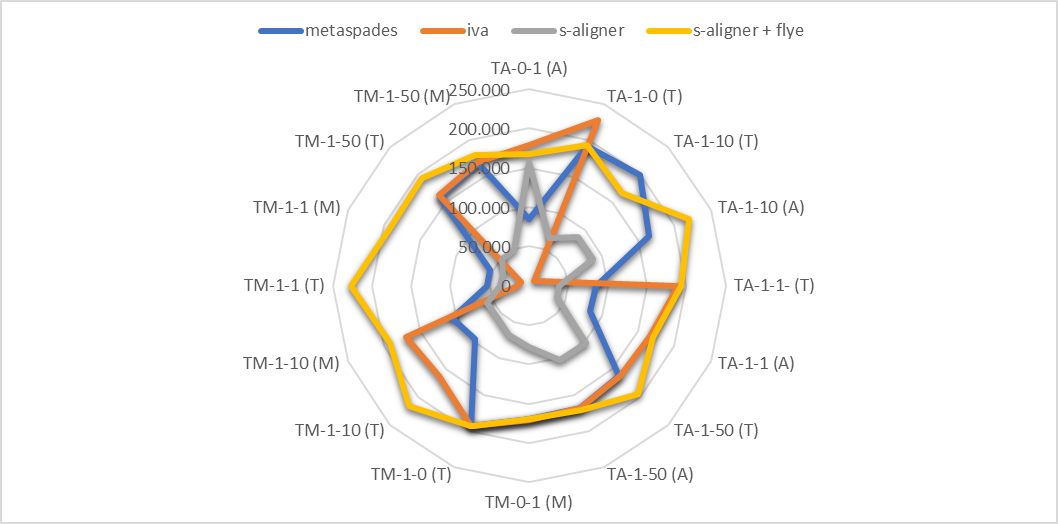
Overall, s-aligner performs on average 110% better than the second-best with the viral benchmark sets analyzed and 64% better with a benchmark set containing samples with extraordinarily large viruses (~250kbp).
All these advantages mean that in a crisis like the one caused by COVID-19, the hundreds of thousands of sequencings being done around the world could make use of cheaper resources to obtain equivalent or superior quality in the results. That could have a significant impact on the management of the crisis.
Halina
It's a shame you don't have a donate button! I'd without a doubt donate tto this fantastic blog! I suppose for now i'll settle for bookmarking and adding your RSS feed to my Google account.I look forward to new updates and will share this blog with my Faacebook group. Talk soon! https://Bandurart.Mystrikingly.com/